Jacobsen Group Research
Mechanism
While the identification of practical and useful catalysts represents an immediate result of our efforts, the broader goal of this work is to lay the foundation for future advances in the design of catalysts and other functional molecules. With the aim of identifying general design principles, we apply the most advanced methods of physical-organic chemistry – including reaction progress kinetic analysis, one- and multidimensional free energy relationship studies, kinetic isotope effect studies, and state-of-the-art computational studies – to elucidate the mechanism of action of effective catalysts. This has provided us with insights into how to devise either improved catalysts for known reactions, or brand-new classes of catalysts for novel applications. The ultimate motivation behind our mechanistic work is the highly ambitious and important dream of rational design of both novel catalysts and other functional small molecules.
Learn about some of the mechanistic work from our lab by clicking on the following topics:
Electronic tuning of chiral catalysts
Attractive non-covalent interactions in small molecule catalysis
Electronic tuning of chiral catalysts
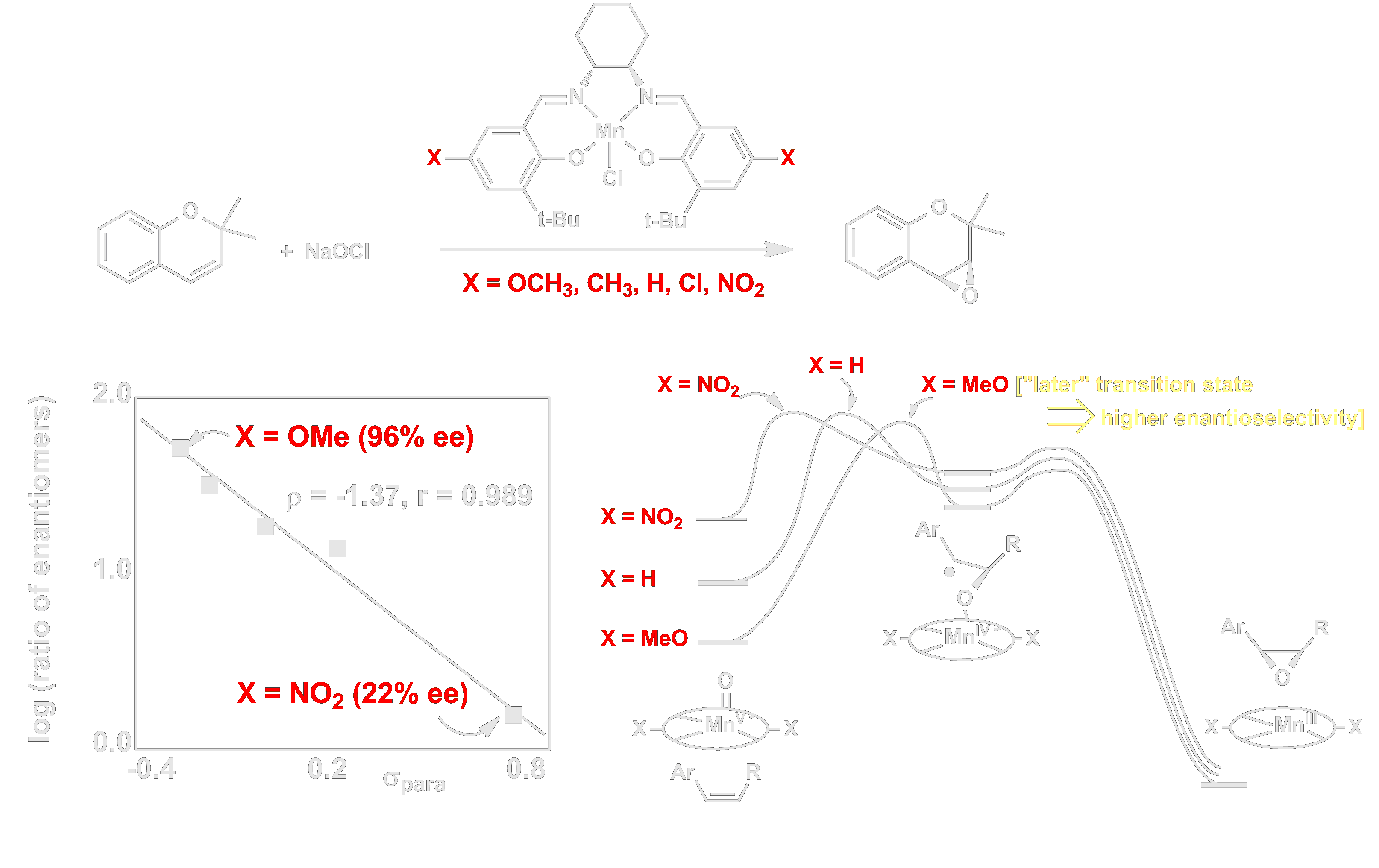
The Mn(salen) epoxidation catalyst developed in our lab was the first monooxygenase model that accomplished highly enantioselective epoxidation, and our lab took full advantage of this new tool for studying the mechanism of oxygen-atom-transfer. In addition, we uncovered several phenomena that hold broader implications in asymmetric catalysis. For example, we found that enantioselectivity in epoxidation can be influenced dramatically by simply tuning the electronic properties of ligand substituents. Through detailed isotope effect studies, we traced these effects to changes in the position of the enantioselectivity-determining transition structure along the reaction coordinate (i.e. the Hammond postulate). This constituted the first direct correlation between the electronic properties of a catalyst and the enantioselectivity it exhibits, and it thus established the concept of electronic tuning of asymmetric catalysts. This has since become a standard tool in the field and been applied successfully by several leading research laboratories.
Catalyst Cooperativity
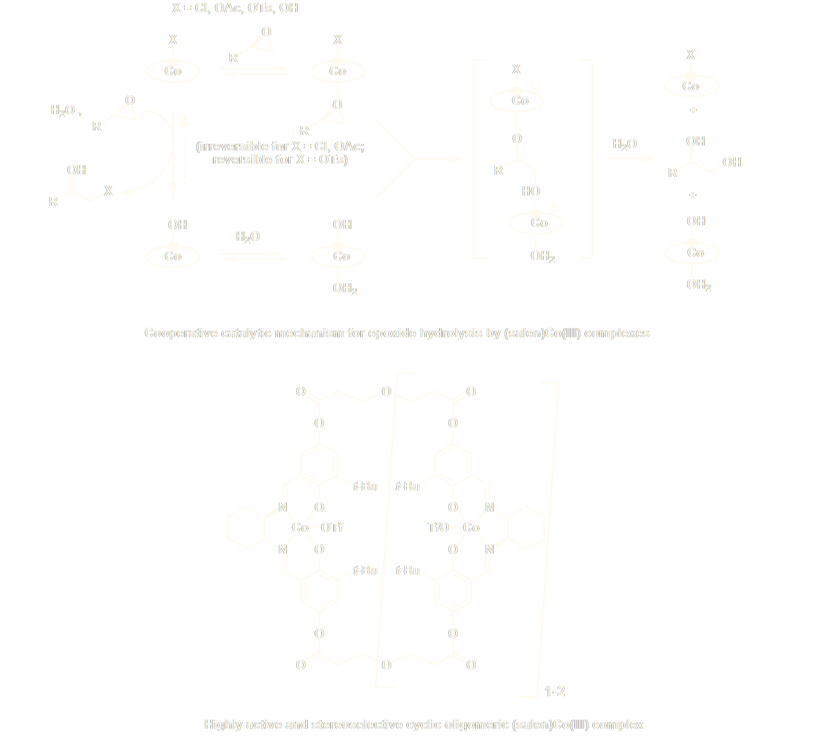
Our group’s mechanistic studies of (salen)metal-catalyzed epoxide-ring-opening reactions have led to important fundamental and practical advances. We established through kinetic and structural studies that these reactions involve cooperative mechanisms where different (salen)metal units activate both the nucleophile and the epoxide, and that the rate- and stereoselectivity-determining step involves a bimetallic transition state in which nucleophile is delivered from one complex to the bound electrophile of another. Based on this insight, we have designed covalently linked multimeric complexes and cyclic oligomeric catalysts where cooperative reactivity is enforced. In particular, the oligomeric (salen)Co catalysts display orders-of-magnitude times faster rates than analogous monomeric catalyst, expanded substrate scope, and higher enantioselectivity. On a broader level, this work helped define a new paradigm of bimetallic catalysis for stereoselective nucleophile-electrophile reactions. We have demonstrated the broader potential of this strategy with some of the first successful examples of cooperative dual asymmetric catalysis, where two different chiral complexes are employed for cooperative activation of reacting partners in synthetically valuable transformations.
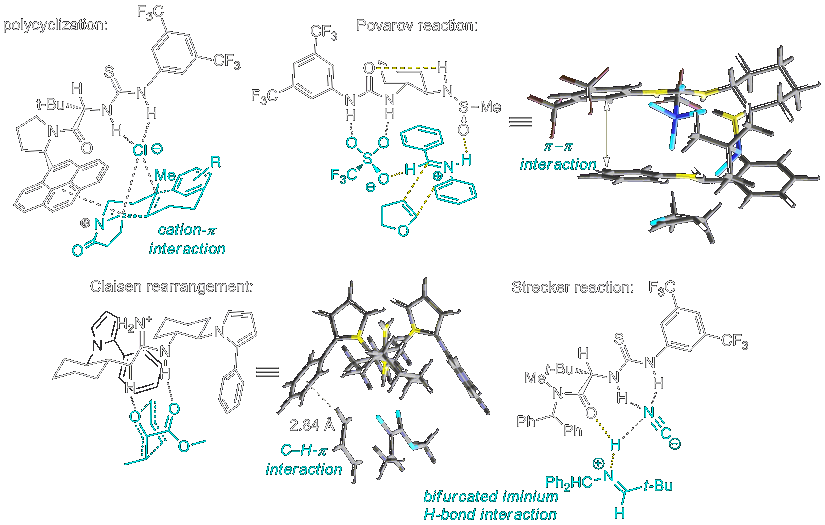
Attractive Non-Covalent Interactions in Small Molecule Catalysis
Transition state-stabilization underlies both rate acceleration and selectivity in enzymes, but the systematic use of attractive interactions has generally not been applied in small molecule asymmetric catalysis prior to our work. Over the past several years, our lab has identified a wide range of simple organocatalytic systems that operate exclusively through non-covalent interactions and induce high levels of enantioselectivity in synthetically important transformations. These small molecules achieve precise control of transition structure geometry through networks of attractive non-covalent interactions (e.g., H-bonding, electrostatic, CH-π). This cooperative model of binding is a defining feature of both biological and synthetic receptors, and also underlies enzymatic catalysis.
Through intensive mechanistic analyses involving state-of-the-art experimental and computational methods, we have probed successfully the mechanisms of stereoinduction in several thiourea-, urea-, and guanidinium-catalyzed enantioselective reactions.